




2. 上海科技大学生命科学与技术学院 上海 201210
2. School of Life Science and Technology, ShanghaiTech University, Shanghai 201210, China
大数据时代,促进科研信息化已成为国家核心竞争力的发展战略之一。为在科研中保持优势和创新力,欧盟、美国、英国相继出台多项政策促进科研信息化发展。我国也于2015 年 8 月由国务院印发《促进大数据发展行动纲要》,以期快速实现“发展科学大数据”的目标。科研信息化的基础设施建设全面加速发展,使超级计算机成为全球科技发展竞争和经济增长的基本工具之一[1]。E 级(Exascale,百亿亿次计算能力)高性能计算机成为世界各国的追求目标之一。近年来,我国高性能计算技术取得了举世瞩目的成就。而在科学研究的整个生命周期中,科研信息化可以促进所有学科的协同、计算或数据密集型研究的创新,推动交叉学科的快速发展。其中,伴随着合理药物设计方法主导的药物研发模式的转变,新药发现过程迎来了一个新的春天,从 2010 年后,美国食品和药物管理局(FDA)批准的新药数量出现了显著增长[2]。尤其 2015 年,FDA 批准了 44 种新药,达到最近 19 年的最高值[3]。
科研信息化基础设施的升级,包括高性能计算机处理器技术的不断更新,新的资源提供模式(网格和云计算)的出现,使得超算技术与计算生物学、计算化学密切结合,同时也为传统的药物发现和计算机辅助药物设计增添了新功能[4],加快了分子动力学模拟和虚拟筛选等的研究进程。在生命科学研究中,云计算已成为未来必不可少的基础设施的重要部分[5]。目前,云计算已被应用到高通量 DNA 测序[6, 7]、新一代测序技术(next-generationsequencing,NGS)[8]和蛋白质组学[9]等的研究中。在生物信息学技术的基础上,联合运用计算机辅助药物设计、结构生物学、化学信息学和网络药理学等学科的研究方法,有助于挖掘已知和发现未知的蛋白-蛋白相互作用。此外,计算技术的合理应用也为在海量的蛋白组学信息中发现可靶性的蛋白-蛋白相互作用提供了新的可能[10]。
1 药物设计方法研究近年来,分子生物学和结构生物学快速发展,阐明了大量生物靶标大分子的三维结构和功能;计算机科学的发展和高性能计算机的出现,又极大地提高了数据计算、分析的速度和精度。在此基础上,基于计算模拟的药物分子设计已作为一种实用化的工具介入到了药物研究的各个环节,并已成为创新药物研究的核心技术之一[11]。我国科研人员在药物设计方法学研究方面也做了大量创新性工作,掌握了一些具有特色和优势的关键核心技术:比如开发的药物靶标预测方法和程序 TarFisDock[12],具有自主知识产权的药物设计软件包 D3Pharm,北京大学来鲁华教授课题组研发的全新药物设计程序 LigBuilder[13, 14],刘晓峰等人发展的基于“反向药效团匹配”策略的靶标预测方法 PharmMapper(并提供了相应的网页服务)[15]等。这些方法已在国际上得到广泛应用,占据了国际生物信息技术开发前沿领域的一席之地。
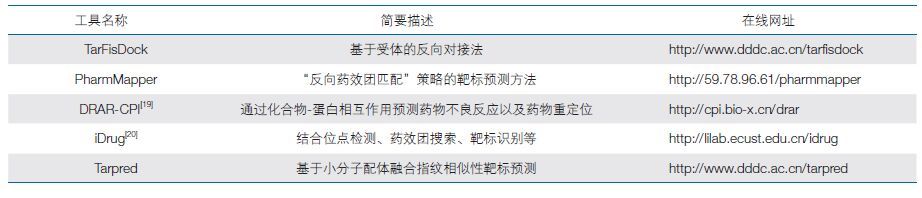
在过去的几十年中,传统的基于特定靶标的药物设计策略在先导化合物发现中起了主导作用,其优点是用更低的成本来获得更高的通量。虽然运用上述策略能够发现高活性高选择性且结构新颖的化合物,但成药性不高,其原因主要是缺少对系统层面药物作用机制的认识及药物的脱靶效应。因此,药物潜在的靶标识别在新药研发过程中具有十分重要的意义,有助于发现类药化合物全新的治疗作用以及一些意想不到的毒副作用,研究人员可据此及时调整研发策略。此外,随着技术的进步和发展,生物活性数据呈指数型增长,传统的方法很难充分利用这些庞大的数据信息。利用相似性融合的思想,建立了一个计算速度快、耗费资源少的基于小分子配体K最近邻(k-NearestNeighbor,KNN)融合指纹相似性的靶标预测模型[16]。通过和近年来靶标预测方面性能较为突出的相似性集成算法(Similarity Ensemble Approach,SEA)[17]进行对比,可以发现 KNN 融合策略要优于 SEA 算法。进一步开发了一款界面友好的免费在线靶标预测工具 Tarpred[18],可以很好地帮助研究人员对小分子可能的治疗靶标和副作用靶标进行预测,以便调整新药研发策略,及时规避风险。众多的工作与成果已经表明,计算机辅助靶标识别作为一种快速且低成本的研究方法,对于药物多靶标作用的预测、药物脱靶效应的发现和小分子调控网络的构建都具有十分重要的意义。下表列举了一些国内研究人员开发的在线靶标预测工具(
除了药物潜在作用靶标预测,发展体系普适性强、计算效率高和计算精度高的配体-受体结合热力学参数(如结合自由能 Δ
在药物设计中,分子对接方法显著加速了先导化合物的发现进程,是目前创新药物研发过程中应用最为广泛的虚拟筛选方法之一[32]。正确的配体-受体结合构象预测和准确的活性预测是分子对接的两个重要目标,二者的实现取决于用于靶标-配体相互作用评价的打分函数。然而,现有的计算方法在靶标-配体相互作用评价方面仍存在着许多问题,包括:(1)无法较好地考虑受体柔性;(2)难以准确描述配体结合过程的溶剂化效应;(3)化合物的活性结合构象识别能力低;(4)靶标-配体结合自由能预测精度差等[23]。针对上述问题,系统地研究和改进了基于知识的靶标-配体相互作用打分函数和计算模型,发展了一系列具有创新性的方法。在考虑受体柔性方面,提出了一种基于统计分析的自适应构象取样方法,可用于构建蛋白质构象变化的马尔科夫状态模型[33];在提高分子对接打分函数性能方面,发展了一种新型的二维氢键统计势,显著提高了蛋白-配体复合物结合构象的识别能力[34];针对在分子对接打分函数中考虑溶剂效应这一难点问题,发展了可以对蛋白质结合腔的水合位点以及结晶水保守性进行预测的统计势方法 wPMF[35]。此外,沈倩诚等人[36]还发展了可以较好地对打分函数的“知识”基础进行扩充的统计势迭代优化方法,通过与包括 Glide 在内的 7 种常见的打分函数进行比较,发现经过优化后的统计势能图物理意义更加清晰,得到的打分函数在结合亲和性预测方面也有了明显的改进。
针对卤键(有机化合物分子中的卤素原子与另一分子中的 O、N、S 等带有部分负电荷的重原子、芳香环结构或 π 键体系之间的吸引作用)在药物设计研究中的重要作用[37],计算模拟了卤键作用的本质,以及与氢键及阳离子 -π 键作用的相互影响,发现它们之间既可以相互协同加强也可以互相拮抗减弱药物与靶标蛋白之间的结合[38]。在此基础上,还探索了卤键对化合物成药性的影响及原因[39],发展了可以用于药物设计的卤键打分函数[40],可快速评价药物与靶标蛋白之间的卤键作用与强度。据此进一步开展了先导化合物结构优化及老药重定位研究,提高了先导化合物的活性,发现了一些老药的新用途信息[41, 42]。这些新型靶标-配体相互作用预测方法对于提高目前合理药物设计在先导化合物发现方面的有效性和成功率具有积极意义。
2 药代动力学模型开发药代动力学性质不良及毒性是造成药物在开发后期失败的重要原因之一。在药物研发早期,开展吸收、分布、代谢、排泄和毒性(ADME/T)实验或计算评价,可以减少人力、物力的浪费,而后者优势尤为明显。ADME/T 预测方法发展和应用研究,已经成为药物发现和优化研究的一个重要研究方向。
伴随着超级计算机的发展,曾经难以实现的众多计算密集型工作得以有效开展。在药物的理化性质与类药性研究方面,通过大规模实验值训练模型,已有多个成功的酸碱离解常数
近年来,随着计算能力和资源的提升,综合考虑代谢[49]、P 糖蛋白(P-glycoprotein,P-gp)和细胞色素 P450(Cytochrome P450,CYP450)酶[50],以及渗透性和溶解性[51]对化合物吸收的影响成为可能,同时也提高了口服生物利用度等预测的准确性。采用小分子与人血清白蛋白(Human Serum Albumin,HSA)之间作用描述符及分子描述符,已有多个关于药物 HSA 结合预测模型[52-54]的报道。在考虑了血浆蛋白结合及脑组织结合的影响,以及利用大数据和随机森林(Random Forest,RF)、支持向量机(Support Vector Machine,SVM)建模等计算方法的基础上,多个高准确率的血脑屏障预测模型得以成功构建[55, 56]。中科院上海药物所联合运用遗传算法(GeneticAlgorithm,GA)与 SVM 来构建药物血脑屏障渗透性定量预测模型,模型在分析发现羧基、极性表面积/氢键形成能力、亲脂性和电荷分布方面有重要作用[57]。此外分子动力学模拟方法预测血脑屏障渗透性参数则为血脑屏障渗透的理论研究指出了新方向[58]。
另一方面,非 P-gp 类跨膜转运体的研究工作也取得了长足进展,包括有机阳离子转运体 2(OCT2)抑制剂、多药和毒物外排转运体 1(MATE1)抑制剂等多种药物转运体抑制剂的发现[59, 60]为预测模型的构建奠定了基础。国内也有利用 GA-CG-SVM 方法成功构建乳腺癌抗性蛋白(BCRP)底物预测模型的报道[61]。中科院上海药物所采用组合药效团策略,成功构建了 OCT2、MATE1 抑制剂组合药效团模型(

|
| 图 1 MATE 1 抑制剂预测模型构建流程 |
CYP 代谢位点预测方面,通过采用柔性分子对接和构象系综考虑 CYP2D6 药物代谢位点柔性,并根据候选位点与催化中心的距离、反应位点的内在反应性等预测代谢位点,取得较好结果[64, 65]。SMARTCyp 软件的出现使得根据分子 2D 结构预测其 CYP3A4 代谢位点成为可能[66],进一步将 SMARTCyp 推广到 CYP1A2、2A6、2B6、2C19、2C8、2C9、2D6 和 2E1 代谢位点的预测,也取得了较好的预测结果[67-69];而采用描述符空间二次抽样和系综方法构建CYP代谢位点预测模型,使模型成功实现了预测与训练集距离较远的分子[70]。针对Ⅱ相代谢反应,中科院上海药物所也有联合使用遗传算法和支持向量机,采用表征活性的量化描述符、分子体积和脂溶性等描述符研究葡萄糖醛酸转移酶(UGTs)的成功报道[71]。
致突变/致癌毒性研究方面,McCarren 等[72]分析了对 Ames 预测模型有重要影响的描述符,并分析了不同数据集对模型的影响。唐赟等[73]采用 SVM、决策树(Decision Tree,DT)、人工神经网络(Artificial NeuralNetwork,ANN)、KNN 和朴素贝叶斯(Naive Bayesian,NB)方法,以及 CDK 分子指纹等为描述符构建了预测模型。中科院上海药物所则使用 Gaston 方法进行致癌警示结构和调节因子的挖掘,发现一些新警示结构[74]。
采用 SVM、DT、RF和NB 方法、递归分割方法以及深度学习方法构建急性毒性预测模型已取得了初步成功[75-77]。而 Drwal 等人[78]发展了一种综合考虑化合物相似性与毒性碎片的急性毒性预测方法,并可根据毒效团预测可能的靶标。中科院上海药物所采用即时学习方法构建了大鼠急性毒性局部预测模型,并通过构建一致性模型提升模型预测性能[76]。Davis 等人[79, 80]构建了 CTD(Comparative Toxicogenomics Database)系统毒理学数据库,为研究人员开展化合物作用机制、疾病机理提供帮助。
随着计算技术的迅猛发展,高质量ADME/T实验数据积累和公布,相关分子机制的阐明,以及新的先进预测算法和抽样方法等的应用,大规模计算已经并将继续深远地影响包括药代动力学在内的众多药物开发过程。而由于技术的不断进步,ADME/T 预测模型的质量将越来越高,并朝实用化迈进,可以乐观地预见,在不远的将来,使用计算方法预判药代动力学性质及毒性反应,并为后期开发提供警示将成为药物设计过程中的重要环节。
3 药物设计方法的应用过去 30 年中,伴随着计算机性能的快速提升和计算机辅助药物设计方法研究的深入,药物设计技术应用领域已由原先的活性化合物发现与优化向上下游拓展深化,逐步应用到包括药物作用靶标发现、药物开发阶段的药效学评价、代谢研究、安全性评价(ADME/T)以及制剂研究等各个新药开发研究领域[11]。此外,计算技术的靶标确证技术的发展、蛋白晶体解析技术的成熟以及众多新方法、新技术的不断涌现使得从药物靶标发现及确证到小分子以及多肽及蛋白相关的药物设计取得了长足进步,也产出了丰硕的成果。尤其是在发现新的生物活性化合物(Hit)等方面有着出色的表现。从应用领域看,药物设计学科近年来的发展方向可大致分为如下3个方面(

|
| 图 2 药物设计学已成为与化学、生物学所齐名的药物研发工具 |
靶标是指存在于组织细胞内与药物相互作用,并赋予药物效应的特定分子。药物作用新靶标的发现以及现有靶标的新功能研究,是小分子探针及原创(First-inclass)药物的源头。化学相似性搜索、数据挖掘、机器学习、反向对接,生物活性谱分析、基于模型的方法以及基于各类分子网络方法的发展为寻找重大疾病具有成药性的新型靶标及开发新的靶向药物提供了契机[81-84]。同时,这些方法的应用也为解释药物的脱靶效应及药物的副作用预测提供了支撑与依据[85, 86]。结合当前国际药物研究中药物靶标发现、功能研究和新分子实体发现一体化趋势和我国药物研发的需求,中科院上海药物所在原有研究的基础上,建立了集靶标发现、功能确证和药物设计一体化的技术平台,成为我国药物设计研究重要的中心之一,对我国药物设计领域的发展作出了突出的贡献,在国内外产生了较大的影响[87-89]。其中针对细菌感染和表观遗传等重要领域,发现了一系列新的候选靶标,并针对发现的潜在药物靶标进行药物发现研究,取得了较好的进展。一系列新靶标、新机制的发现深化了对于生命调控过程的理解,也拓展了药物作用位点和机制,为新型高活低毒药物的诞生提供了可能[90, 91]。
靶标发现的另一个重要发展趋势是,基于生物信息学、系统生物学分析发现疾病相关的新的信号转导通路和生物调控网络,分析潜在靶标在生物网络中所处的位置及其对网络功能的影响,为深入理解疾病及其药物治疗奠定基础。癌症、糖尿病及心血管疾病等复杂疾病的自身特点使得传统的针对单一靶标药物难以发挥效果达成治疗目标。近年,Pf izer 的研究人员与 Allen,B. K.及其合作者先后通过基于结构的药物设计方法以及结合机器学习的大规模虚拟筛选发现了作用于癌症信号通路的双靶标抑制剂,确定了使用计算机方法进行基于信号通路药物研究的可行性[92, 93]。而构建疾病相关生物分子网络,分析网络动力学性质,寻找网络敏感位点则为药物发现提供了新方向[94]。通过针对一些典型的生物功能模块的深入研究,理解其网络核心拓扑结构、参数限制、关键节点搜寻及调控机制,中科院上海药物所成功构建了包括炎-癌转变网络在内的数个复杂生物网络模型,从系统的角度理解蛋白调控网络问题,为复杂疾病网络的化学干预预测提供了新思路[95]。
传统意义上靶标确证及功能研究主要通过实验手段来进行,耗时费力,复杂体系不易操作。计算生物学方法作为一种生物学研究工具,特别是分子动力学模拟方法通过长时间的模拟生物大分子靶标的构象变化,可以从微观层面研究其功能,有效地弥补了实验方法的不足。近年来,G 蛋白偶联受体(Guanosine-bindingProtein Coupled Receptor,GPCR)和离子通道蛋白作为一类重要的潜在药物靶标,受到越来越多的重视。其动态构象研究以及离子通道,特别是电压门控离子(Kv)通道和酸敏感离子通道的门控机制研究取得了较大进展。中科院上海药物所应用计算生物学方法率先在全长 B 类 GPCR 动态构象研究领域取得突破[96],首次揭示全长 B 类 GPCR 的 ECD 和跨膜区具有两种相对构象,推动了 B 类 GPCR 的结构功能关系研究,也为设计基于新机制的 B类 GPCR 的功能小分子提供了重要信息。美国科学家通过计算生物学方法研究了 Kv1.2/Kv2.1 通道从开放到静息再到开放的状态转变,完整描述了 Kv 通道的开放关闭过程[97]。针对另一类重要的药物靶标——酸敏感质子通道(ASIC),中科院上海药物所和中科院神经科学所研究人员合作利用计算生物学方法率先研究了 ASIC1 的动态行为[98, 99](

|
| 图 3 Kv 通道、ASIC 通道和 GPCR 的配体调控位点示意图 |
小分子对跨膜蛋白动态构象的调控是跨膜蛋白研究领域的另一个重要关注点。研究组成细胞膜的磷脂和胆固醇等脂类小分子与跨膜蛋白作用和调控跨膜蛋白的生理及病理功能的分子机制,是当前离子通道结构功能关系研究的重要研究方向。中科院上海药物所通过大规模分子动力学模拟与实验研究结合,阐述了 PIP2 与 KCNQ2 通道结合的动态过程[100]。在此基础上,通过更长时间(1μs)分子动力学模拟发现了 PIP2 调控 KCNQ2 通道的新机制[101]。此外,通过长时间 MD 模拟发现磷脂对同源蛋白的差异化调控不局限于离子通道。这些研究为在分子水平上探索磷脂分子调控膜蛋白提供了结构基础和新见解,同时为理解胆固醇分子调控 GPCR 的功能多样性提供了结构基础[102, 103]。
近年来,随着新的候选靶标不断涌现,在没有现成活性分子参考的情况下,药物设计技术在新靶标第一代活性分子发现中起到了不可替代的作用。从第一代活性分子开始,结合药物设计技术进行结构优化,从而实现从无到有、从有到精的分子实体发现。基于结构和基于配体的药物设计技术在整个新药研发阶段中占有不可替代的独特地位,为开发选择性高、活性好的先导化合物提供了重要的技术支撑。利用基于结构的药物设计(Structure BasedDrug Design,SBDD)技术,根据同源模建或晶体结构,使用经典分子对接方法进行虚拟筛选现已成为富集活性先导化合物、加速药物发现过程的有效手段[23]。众多实验结果已经证明,计算机辅助药物设计在包括癌症、代谢疾病、神经系统疾病、心血管疾病、呼吸系统疾病等众多疾病治疗领域均有着杰出的应用前景[104-110],屡有针对其关键蛋白的作用方式独特兼具杰出活性与靶标特异性的优异活性化合物实体被发现[111-113]。近年,中科院上海药物所通过与芝加哥大学、艾默里大学等单位合作,利用虚拟筛选策略,发现了高活性高选择性小分子DC-AC50,可同时靶向两种铜伴侣蛋白Atox1和CCS,选择性调控铜离子转运,从而特异性抑制肿瘤细胞增殖,且在多种动物实验中表现出良好的抗肿瘤活性[114]。目前,DC-AC50 相关成果已经实现转化。
与 GPCR 和激酶等靶标不同,电压门控通道是被电压激活,没有明确的常规内源性配体结合口袋。确证激动剂的作用位点是电压门控通道研究领域的难点之一,通过基于结构的药物设计发现电压门控通道激动剂也进而面临很大挑战。计算生物学研究直接推动了基于动态构象的药物设计,使得这些药物靶标的理性药物设计成为可能。KCNQ2 是癫痫相关的一类电压门控钾离子通道,其通道激动剂被证实可以缓解人类的癫痫症状。中科院上海药物所通过综合运用动力学模拟、分子对接、定点突变和电生理测试等方法,发现了一个位于通道门控电荷通路(gating charge pathway)中的激动剂结合口袋[115]并针对该口袋开展基于结构的药物设计。经电生理测试确认了 9 个 KCNQ2 新激动剂,其中两个在两类动物模型中表现出优异的抗癫痫活性。该研究为发现离子通道调制剂结合口袋提供了成功的案例,并且首次实现了“基于结构的电压门控钾离子通道激动剂发现”,为离子通道药物研究领域的一个重要进展。
蛋白-蛋白相互作用(Protein-Protein Interaction,PPI)是许多生命过程得以顺利完成的基础,同时也是一类重要的药物作用靶标,其应用前景广泛、市场潜力巨大。相比于传统的高通量筛选,使用基于片段的药物设计与改进的虚拟筛选方法在寻找 PPI 抑制剂方面更为有效[116]。靶向蛋白-蛋白相互作用药物设计的要点是寻找作用界面上的关键氨基酸残基(hot spots)[117]。作为一个新兴的研究领域,近年来,蛋白-蛋白相互作用研究得到越来越多的关注[118, 119]。目前,已被批准用于临床的基于多肽设计的药物超过 50 种。国内也已报道了使用计算模拟与实验验证相结合方式的包括电压门控的钾离子通道(Kv1.3)抑制性多肽设计等多个成功案例[120],中科院上海药物所也有通过对表观遗传调控蛋白复合物 PRC2 的界面性质研究发现第一个靶向 EED-EZH2 复合物界面的小分子抑制剂的成功案例[121]。
目前,基于网络的药物设计还处在起步阶段,已构建的分子网络模型数量、规模有限,对靶点/蛋白间相互影响的认识也较为肤浅。但基于生物体自身调控网络的复杂性,从系统层面对疾病与药物的理解至关重要,基于调控网络的药物设计方法也是药物设计领域的一大方向。随着对于疾病网络层面的理解的加深,分子网络模型正在不断发展与完善,可以预见,在不远的将来,基于对疾病的系统理解,癌症、神经系统疾病等复杂疾病的多靶标药物联用治疗将成为可能。而另一方面,由于蛋白-蛋白相互作用界面的关键残基难以预测和模拟,现阶段作用于蛋白-蛋白相互作用的小分子抑制剂数量还相对较少,如何有效地模拟和预测蛋白-蛋白作用过程仍是药物设计学领域的一大难题。经典的小分子虚拟筛选方式似乎并不适用于蛋白-蛋白作用界面抑制剂的筛选[122],而如何捕获蛋白-蛋白相互作用反应过程的动态构型、使用柔性蛋白构象进行分子库虚筛则是蛋白-蛋白相互作用小分子设计的先决条件,也是药物设计学发展的重要方向。
4 总结综上所述,由于药物研发流程的复杂性,近年来大量自动化设备的使用,研发产生的海量数据也有着不同类型,这标志着医药研发大数据时代的来临。基于研发大数据时代的建模与模拟,需要建立企业级的研发信息高速公路。如今,制药公司和学术机构的合作也更为紧密,甚至形成了所谓的“开源药物”研发模式。另外,研发领域出现了众多移动应用,并与云端形成合作平台,如全球利用云技术进行罕见病的合作研究,研发模式与互联网时代同进化。其中最为关键的就是整合不同来源和结构的数据,解析不同来源数据之间的关联,对原始数据进行不同维度的数据解析。中科院上海药物所已经成功构建了包涵情报信息系统、GRP 实验管理系统和 GLP 新药申报系统等多项内容的“新药研发集成化平台”,初步形成完整的新药研发标准化流程管理系统。
信息技术改变了药物研发模式。药物研发信息化可以帮助我们提高效率、降低成本,加快研发进程,进行风险控制,并提升研发价值和创新能力,使研发业务真正向智能化迈进,让药物研发各参与者摆脱盲人摸象的状态。
| [1] | Ge H, Wang Y, Li C, et al. Molecular dynamics-based virtual screening: Accelerating the drug discovery process by highperformance computing. J. Chem. Inf. Model, 2013, 53(10):2757-2764. |
| Cited By in Cnki | |
| [2] | Mullard A. 2014 FDA drug approvals. Nat. Rev. Drug Discov.,2015, 14 (2): 77-81. |
| Cited By in Cnki | |
| [3] | Booth B L. This time may be different. Nat. Biotechnol., 2016,34 (1): 25-30. |
| Cited By in Cnki | |
| [4] | Pitera J W. Current developments in and importance of highperformance computing in drug discovery. Curr. Opin. Drug Disc., 2009, 12 (3): 388-396. |
| Cited By in Cnki | |
| [5] | Stein L D. The case for cloud computing in genome informatics. Genome Biol., 2010, 11 (5): 79-82. |
| Cited By in Cnki | |
| [6] | Jourdren L, Bernard M, Dillies M-A, et al. Eoulsan: A cloud computing-based framework facilitating high throughput sequencing analyses. Bioinformatics, 2012, 28 (11): 1542-1543. |
| Cited By in Cnki | |
| [7] | Moghadam B T, Alvarsson J, Holm M, et al. Scaling predictive modeling in drug development with cloud computing. J. Chem. Inf. Model, 2015, 55 (1): 19-25. |
| Cited By in Cnki | |
| [8] | Kwon T, Yoo W G, Lee W-J, et al. Next-generation sequencing data analysis on cloud computing. Genes Genom., 2015, 37 (6):489-501. |
| Cited By in Cnki | |
| [9] | Mohammed Y, Mostovenko E, Henneman A A, et al. Cloud parallel processing of tandem mass spectrometry based proteomics data. J. Proteome Res., 2012, 11(10): 5101-5108. |
| Cited By in Cnki | |
| [10] | Milroy L-G, Grossmann T N, Hennig S, et al. Modulators of protein-protein interactions. Chem. Rev., 2014, 114(9): 4695-4748. |
| Cited By in Cnki | |
| [11] | Zheng M Y, Liu X, Xu Y, et al. Computational methods for drug design and discovery: Focus on china. Trends Pharmacol. Sci.,2013, 34(10): 549-559. |
| Cited By in Cnki | |
| [12] | Li H L, Gao Z T, Kang L, et al. A web server for identifying drug targets with docking approach. Nucleic Acids Res., 2006, 34(SI):219-224. |
| Cited By in Cnki | |
| [13] | Wang R X, Gao Y, Lai L H. Ligbuilder: A multi-purpose program for structure-based drug design. J. Mol. Model, 2000, 6(7-8):498-516. |
| Cited By in Cnki | |
| [14] | Yuan Y X, Pei J F, Lai L H, et al. Ligbuilder 2: A practical de novo drug design approach. J. Chem. Inf. Model, 2011, 51(5):1083-1091. |
| Cited By in Cnki | |
| [15] | Liu X F, Ouyang S S, Yu B, et al. Pharmmapper server: A web server for potential drug target identification using pharmacophore mapping approach. Nucleic Acids Res., 2010,38(2): 609-614. |
| Cited By in Cnki | |
| [16] | Liu X, Xu Y, Li S S, et al. In silico target fishing: Addressing a "big data" problem by ligand-based similarity rankings with data fusion. J. Cheminformatics, 2014, 6(1): 33-33. |
| Cited By in Cnki | |
| [17] | Keiser M J, Roth B L, Armbruster B N, et al. Relating protein pharmacology by ligand chemistry. Nat. Biotechnol., 2007,25(2): 197-206. |
| Cited By in Cnki | |
| [18] | Liu X, Gao Y, Peng J L, et al. Tarpred: A web application for predicting therapeutic and side effect targets of chemical compounds. Bioinformatics, 2015, 31(12): 2049-2051. |
| Cited By in Cnki | |
| [19] | Luo H, Chen J, Shi L M, et al. Drar-cpi: A server for identifying drug repositioning potential and adverse drug reactions via the chemical-protein interactome. Nucleic Acids Res., 2011, 39(2):492-498. |
| [20] | Wang X, Chen H P, Yang F, et al. Idrug: A web-accessible and interactive drug discovery and design platform. J. Cheminformatics, 2014, 6(28): 205-230. |
| [21] | Huang S Y, Grinter S Z, Zou X. Scoring functions and their evaluation methods for protein-ligand docking: Recent advances and future directions. Phys. Chem. Chem. Phys., 2010, 12(40):12899-12908. |
| Cited By in Cnki | |
| [22] | Singh N, Warshel A. Absolute binding free energy calculations: On the accuracy of computational scoring of protein-ligand interactions. Proteins, 2010, 78(7): 1705-1723. |
| Cited By in Cnki | |
| [23] | Kitchen D B, Decornez H, Furr J R, et al. Docking and scoring in virtual screening for drug discovery: Methods and applications. Nat. Rev. Drug Discov., 2004, 3(11): 935-949. |
| Cited By in Cnki | |
| [24] | Aqvist J, Mowbray S L. Sugar recognition by a glucose/ galactose receptor-evaluation of binding energetics from molecular-dynamics simulations. J. Biol. Chem., 1995, 270(17):9978-9981. |
| Cited By in Cnki | |
| [25] | Aqvist J, Medina C, Samuelsson J E. New method for predicting binding-affinity in computer-aided drug design. Protein Eng.,1994, 7(3): 385-391. |
| Cited By in Cnki | |
| [26] | Aqvist J. Long-range electrostatic effects on peptide folding. FEBS Lett., 1999, 457(3): 414-418. |
| Cited By in Cnki | |
| [27] | Wang W, Donini O, Reyes C M, et al. Biomolecular simulations: Recent developments in force fields, simulations of enzyme catalysis, protein-ligand, protein-protein, and protein-nucleic acid noncovalent interactions. Annu. Rev. Biophys. Biomol. Struct., 2001, 30(30): 211-243. |
| Cited By in Cnki | |
| [28] | Adcock S A, Mccammon J A. Molecular dynamics: Survey of methods for simulating the activity of proteins. Chem. Rev.,2006, 106(5): 1589-1615. |
| Cited By in Cnki | |
| [29] | Bai F, Xu Y, Chen J, et al. Free energy landscape for the binding process of huperzine a to acetylcholinesterase. Proc. Natl. Acad. Sci. USA, 2013, 110(11): 4273-4278. |
| Cited By in Cnki | |
| [30] | Buch I, Giorgino T, De Fabritiis G. Complete reconstruction of an enzyme-inhibitor binding process by molecular dynamics simulations. Proc. Natl. Acad. Sci. USA, 2011, 108(25): 10184-10189. |
| Cited By in Cnki | |
| [31] | Okazaki K-I, Takada S. Dynamic energy landscape view of coupled binding and protein conformational change: Induced-fit versus population-shift mechanisms. Proc. Natl. Acad. Sci. USA, 2008, 105(32): 11182-11187. |
| Cited By in Cnki | |
| [32] | Shoichet B K. Virtual screening of chemical libraries. Nature,2004, 432(7019): 862-865. |
| Cited By in Cnki | |
| [33] | Li S S, Xiong B, Xu Y, et al. Mechanism of the all-alpha to allbeta conformational transition of rfah-ctd: Molecular dynamics simulation and markov state model. J. Chem. Theory Comput.,2014, 10(6): 2255-2264. |
| Cited By in Cnki | |
| [34] | Zheng M Y, Xiong B, Luo C, et al. Knowledge-based scoring functions in drug design: 3. A two-dimensional knowledge-based hydrogen-bonding potential for the prediction of protein-ligand interactions. J. Chem. Inf. Model, 2011, 51(11): 2994-3004. |
| Cited By in Cnki | |
| [35] | Zheng M Y, Li Y L, Xiong B, et al. Water pmf for predicting the properties of water molecules in protein binding site. J. Comput. Chem., 2013, 34(7): 583-592. |
| Cited By in Cnki | |
| [36] | Shen Q C, Xiong B, Zheng M Y, et al. Knowledge-based scoring functions in drug design: 2. Can the knowledge base be enriched?. J. Chem. Inf. Model, 2011, 51(2): 386-397. |
| Cited By in Cnki | |
| [37] | Lu Y X, Shi T, Wang Y, et al. Halogen bonding-a novel interaction for rational drug design?. J. Med. Chem., 2009,52(9): 2854-2862. |
| Cited By in Cnki | |
| [38] | Lu Y X, Wang Y, Xu Z J, et al. C-x center dot center dot center dot h contacts in biomolecular systems: How they contribute to protein-ligand binding affinity. J. Phys. Chem. B, 2009, 113(37):12615-12621. |
| Cited By in Cnki | |
| [39] | Xu Z J, Yang Z, Liu Y T, et al. Halogen bond: Its role beyond drug-target binding affinity for drug discovery and development. J. Chem. Inf. Model, 2014, 54(1): 69-78. |
| Cited By in Cnki | |
| [40] | Liu Y T, Xu Z J, Yang Z, et al. A knowledge-based halogen bonding scoring function for predicting protein-ligand interactions. J. Mol. Model, 2013, 19(11): 5015-5030. |
| Cited By in Cnki | |
| [41] | Xu Z J, Liu Z, Chen T, et al. Utilization of halogen bond in lead optimization: A case study of rational design of potent phosphodiesterase type 5 (pde5) inhibitors. J. Med. Chem., 2011,54(15): 5607-5611. |
| Cited By in Cnki | |
| [42] | Ren J, He Y, Chen W Y, et al. Thermodynamic and structural characterization of halogen bonding in protein-ligand interactions: A case study of pde5 and its inhibitors. J. Med. Chem., 2014, 57(8): 3588-3593. |
| Cited By in Cnki | |
| [43] | Settimo L, Bellman K, Knegtel R M A. Comparison of the accuracy of experimental and predicted pka values of basic and acidic compounds. Pharm. Res., 2014, 31(4): 1082-1095. |
| Cited By in Cnki | |
| [44] | Fraczkiewicz R, Lobell M, Goeller A H, et al. Best of both worlds: Combining pharma data and state of the art modeling technology to improve in silico pk(a) prediction. J. Chem. Inf. Model, 2015, 55(2): 389-397. |
| Cited By in Cnki | |
| [45] | Lusci A, Pollastri G, Baldi P. Deep architectures and deep learning in chemoinformatics: The prediction of aqueous solubility for drug-like molecules. J. Chem. Inf. Model, 2013,53(7): 1563-1575. |
| Cited By in Cnki | |
| [46] | Raevsky O A, Polianczyk D E, Grigorev V Y, et al. In silico prediction of aqueous solubility: A comparative study of local and global predictive models. Mol. Inf., 2015, 34(6-7): 417-430. |
| Cited By in Cnki | |
| [47] | Bickerton G R, Paolini G V, Besnard J, et al. Quantifying the chemical beauty of drugs. Nat. Chem., 2012, 4(2): 90-98. |
| Cited By in Cnki | |
| [48] | Ritchie T J, Macdonald S J F. How drug-like are ‘ugly’ drugs: Do drug-likeness metrics predict adme behaviour in humans?. Drug Discov. Today, 2014, 19(4): 489-495. |
| Cited By in Cnki | |
| [49] | Tian S, Li Y Y, Wang J M, et al. Adme evaluation in drug discovery. 9. Prediction of oral bioavailability in humans based on molecular properties and structural fingerprints. Mol. Pharm.,2011, 8(3): 841-851. |
| Cited By in Cnki | |
| [50] | Xu X, Zhang W X, Huang C, et al. A novel chemometric method for the prediction of human oral bioavailability. Int. J. Mol. Sci.,2012, 13(6): 6964-6982. |
| Cited By in Cnki | |
| [51] | Newby D, Freitas A A, Ghafourian T. Decision trees to characterise the roles of permeability and solubility on the prediction of oral absorption. Eur. J. Med. Chem., 2015, 90: 751-765. |
| Cited By in Cnki | |
| [52] | Li H Y, Chen Z X, Xu X J, et al. Predicting human plasma protein binding of drugs using plasma protein interaction qsar analysis (ppi-qsar). Biopharm. Drug Dispos., 2011, 32(6): 333-342. |
| Cited By in Cnki | |
| [53] | Chen L J, Chen X. Results of molecular docking as descriptors to predict human serum albumin binding affinity. J. Mol. Graph.,2012, 33: 35-43. |
| Cited By in Cnki | |
| [54] | Lexa K W, Dolghih E, Jacobson M P. A structure-based model for predicting serum albumin binding. PLoS One, 2014, 9(4): e93323. |
| Cited By in Cnki | |
| [55] | Martins I F, Teixeira A L, Pinheiro L, et al. A bayesian approach to in silico blood-brain barrier penetration modeling. J. Chem. Inf. Model, 2012, 52(6): 1686-1697. |
| Cited By in Cnki | |
| [56] | Lanevskij K, Dapkunas J, Juska L, et al. Qsar analysis of bloodbrain distribution: The influence of plasma and brain tissue binding. J. Pharm. Sci., 2011, 100(6): 2147-2160. |
| Cited By in Cnki | |
| [57] | Zhang D Q, Xiao J F, Zhou N N, et al. A genetic algorithm based support vector machine model for blood-brain barrier penetration prediction. Biomed Res. Int., 2015, 2015: 292683. |
| Cited By in Cnki | |
| [58] | Carpenter T S, Kirshner D A, Lau E Y, et al. A method to predict blood-brain barrier permeability of drug-like compounds using molecular dynamics simulations. Biophys. J., 2014, 107(3): 630-641. |
| Cited By in Cnki | |
| [59] | Kido Y, Matsson P, Giacomini K M. Profiling of a prescription drug library for potential renal drug-drug interactions mediated by the organic cation transporter 2. J. Med. Chem., 2011, 54(13):4548-4558. |
| Cited By in Cnki | |
| [60] | Wittwer M B, Zur A A, Khuri N, et al. Discovery of potent, selective multidrug and toxin extrusion transporter 1 (mate1, slc47a1) inhibitors through prescription drug profiling and computational modeling. J. Med. Chem., 2013, 56(3): 781-795. |
| Cited By in Cnki | |
| [61] | Zhong L, Ma C Y, Zhang H, et al. A prediction model of substrates and non-substrates of breast cancer resistance protein (bcrp) developed by ga-cg-svm method. Comput. Biol. Med.,2011, 41(11): 1006-1013. |
| Cited By in Cnki | |
| [62] | Xu Y, Liu X, Li S S, et al. Combinatorial pharmacophore modeling of organic cation transporter 2 (oct2) inhibitors: Insights into multiple inhibitory mechanisms. Mol. Pharm.,2013, 10(12): 4611-4619. |
| Cited By in Cnki | |
| [63] | Xu Y, Liu X, Wang Y L, et al. Combinatorial pharmacophore modeling of multidrug and toxin extrusion transporter 1 inhibitors: A theoretical perspective for understanding multiple inhibitory mechanisms. Sci. Rep., 2015, (5): 13684. |
| Cited By in Cnki | |
| [64] | Li J, Schneebeli S T, Bylund J, et al. Idsite: An accurate approach to predict p450-mediated drug metabolism. J. Chem. Theory Comput., 2011, 7(11): 3829-3845. |
| Cited By in Cnki | |
| [65] | Moors S L C, Vos A M, Cummings M D, et al. Structure-based site of metabolism prediction for cytochrome p450 2d6. J. Med. Chem., 2011, 54(17): 6098-6105. |
| Cited By in Cnki | |
| [66] | Rydberg P, Gloriam D E, Zaretzki J, et al. Smartcyp: A 2d method for prediction of cytochrome p450-mediated drug metabolism. ACS Med. Chem. Lett., 2010, 1(3): 96-100. |
| Cited By in Cnki | |
| [67] | Rydberg P, Olsen L. Ligand-based site of metabolism prediction for cytochrome p450 2d6. ACS Med. Chem. Lett., 2012, 3(1):69-73. |
| Cited By in Cnki | |
| [68] | Liu R F, Liu J, Tawa G, et al. 2d smartcyp reactivity-based site of metabolism prediction for major drug-metabolizing cytochrome p450 enzymes. J. Chem. Inf. Model, 2012, 52(6): 1698-1712. |
| Cited By in Cnki | |
| [69] | Zaretzki J, Rydberg P, Bergeron C, et al. Rs-predictor models augmented with smartcyp reactivities: Robust metabolic regioselectivity predictions for nine cyp isozymes. J. Chem. Inf. Model, 2012, 52(6): 1637-1659. |
| Cited By in Cnki | |
| [70] | Tyzack J D, Mussa H Y, Williamson M J, et al. Cytochrome p450 site of metabolism prediction from 2d topological fingerprints using gpu accelerated probabilistic classifiers. J. Cheminformatics, 2014, 6(2), 29. |
| Cited By in Cnki | |
| [71] | Peng J L, Lu J, Shen Q C, et al. In silico site of metabolism prediction for human ugt-catalyzed reactions. Bioinformatics,2014, 30(3): 398-405. |
| Cited By in Cnki | |
| [72] | Mccarren P, Springer C, Whitehead L. An investigation into pharmaceutically relevant mutagenicity data and the influence on ames predictive potential. J. Cheminformatics, 2011,3(22):12264-12269. |
| Cited By in Cnki | |
| [73] | Xu C Y, Cheng F X, Chen L, et al. In silico prediction of chemical ames mutagenicity. J .Chem. Inf. Model, 2012, 52(11):2840-2847. |
| Cited By in Cnki | |
| [74] | Wang Y, Lu J, Wang F, et al. Estimation of carcinogenicity using molecular fragments tree. J. Chem. Inf. Model, 2012, 52(8):1994-2003. |
| Cited By in Cnki | |
| [75] | Li X, Chen L, Cheng F X, et al. In silico prediction of chemical acute oral toxicity using multiclassification methods. J. Chem. Inf. Model, 2014, 54(4): 1061-1069. |
| Cited By in Cnki | |
| [76] | Lu J, Peng J L, Wang J N, et al. Estimation of acute oral toxicity in rat using local lazy learning. J. Cheminformatics, 2014, 6(1):334-336. |
| Cited By in Cnki | |
| [77] | Wang S C, Li Y Y, Wang J M, et al. Admet evaluation in drug discovery. 12. Development of binary classification models for prediction of herg potassium channel blockage. Mol. Pharm.,2012, 9(4): 996-1010. |
| Cited By in Cnki | |
| [78] | Drwal M N, Banerjee P, Dunkel M, et al. Protox: A web server for the in silico prediction of rodent oral toxicity. Nucleic Acids Res., 2014, 42(W1): 53-58. |
| Cited By in Cnki | |
| [79] | Davis A P, Murphy C G, Johnson R, et al. The comparative toxicogenomics database: Update 2013. Nucleic Acids Res,2013, 41(D1): 104-114. |
| Cited By in Cnki | |
| [80] | Kongsbak K, Hadrup N, Audouze K, et al. Applicability of computational systems biology in toxicology. Basic Clin. Pharmacol. Toxicol., 2014, 115(1): 45-49. |
| Cited By in Cnki | |
| [81] | Csermely P, Korcsmaros T, Kiss H J M, et al. Structure and dynamics of molecular networks: A novel paradigm of drug discovery a comprehensive review. Pharmacol. Ther., 2013,138(3): 333-408. |
| Cited By in Cnki | |
| [82] | Harrold J M, Ramanathan M, Mager D E. Network-based approaches in drug discovery and early development. Clin. Pharmacol. Ther., 2013, 94(6): 651-658. |
| Cited By in Cnki | |
| [83] | Wang L R, Ma C, Wipf P, et al. Targethunter: An in silico target identification tool for predicting therapeutic potential of small organic molecules based on chemogenomic database. AAPS J.,2013, 15(2): 395-406. |
| Cited By in Cnki | |
| [84] | Yizhak K, Gabay O, Cohen H, et al. Model-based identification of drug targets that revert disrupted metabolism and its application to ageing. Nat. Commun., 2013, 4: 1-11. |
| Cited By in Cnki | |
| [85] | Lounkine E, Keiser M J, Whitebread S, et al. Large-scale prediction and testing of drug activity on side-effect targets. Nature, 2012, 486(7403): 361-367. |
| Cited By in Cnki | |
| [86] | Schenone M, Dancik V, Wagner B K, et al. Target identification and mechanism of action in chemical biology and drug discovery. Nat. Chem. Biol., 2013, 9(4): 232-240. |
| Cited By in Cnki | |
| [87] | Chen S J, Wang Y L, Zhou W, et al. Identifying novel selective non-nucleoside DNA methyltransferase 1 inhibitors through docking-based virtual screening. J. Med. Chem., 2014, 57(21):9028-9041. |
| Cited By in Cnki | |
| [88] | Zhang J, Liu H C, Zhu K K, et al. Antiinfective therapy with a small molecule inhibitor of staphylococcus aureus sortase. Proc. Natl. Acad. Sci. USA, 2014, 111(37): 13517-13522. |
| Cited By in Cnki | |
| [89] | Huang Y, Yan J L, Li Q, et al. Meclofenamic acid selectively inhibits fto demethylation of m(6)a over alkbh5. Nucleic Acids Res., 2015, 43(1): 373-384. |
| Cited By in Cnki | |
| [90] | Chen Z, Wang X, Zhu W P, et al. Acenaphtho 1,2-b pyrrolebased selective fibroblast growth factor receptors 1 (fgfr1) inhibitors: Design, synthesis, and biological activity. J. Med. Chem., 2011, 54(11): 3732-3745. |
| Cited By in Cnki | |
| [91] | Sun W Y, Xie Z Q, Liu Y F, et al. Jx06 selectively inhibits pyruvate dehydrogenase kinase pdk1 by a covalent cysteine modification. Cancer Res., 2015, 75(22): 4923-4936. |
| Cited By in Cnki | |
| [92] | Cui J J, Tran-Dube M, Shen H, et al. Structure based drug design of crizotinib (pf-02341066), a potent and selective dual inhibitor of mesenchymal-epithelial transition factor (c-met) kinase and anaplastic lymphoma kinase (alk). J. Med. Chem., 2011, 54(18):6342-6363. |
| Cited By in Cnki | |
| [93] | Allen B K, Mehta S, Ember S W J, et al. Large-scale computational screening identifies first in class multitarget inhibitor of egfr kinase and brd4. Sci. Rep., 2015, 5: 16924. |
| Cited By in Cnki | |
| [94] | Yang K, Bai H J, Qi O Y, et al. Finding multiple target optimal intervention in disease-related molecular network. Mol. Syst. Biol., 2008, 4(1): 228. |
| Cited By in Cnki | |
| [95] | Lu J Y, Zeng H L, Liang Z J, et al. Network modelling reveals the mechanism underlying colitis-associated colon cancer and identifies novel combinatorial anti-cancer targets. Sci. Rep.,2015, 5: 2045-2322. |
| Cited By in Cnki | |
| [96] | Yang L L, Yang D H, De Graaf C, et al. Conformational states of the full-length glucagon receptor. Nat. Commun., 2015, 6(7859):2708-2713. |
| Cited By in Cnki | |
| [97] | Jensen M O, Jogini V, Borhani D W, et al. Mechanism of voltage gating in potassium channels. Science, 2012, 336(6078): 229-233. |
| Cited By in Cnki | |
| [98] | Yang H Y, Yu Y, Li W G, et al. Inherent dynamics of the acidsensing ion channel 1 correlates with the gating mechanism. Plos Biol., 2009, 7(7): e1000151. |
| Cited By in Cnki | |
| [99] | Yang H Y, Yu Y, Li W G, et al. Conformational sampling on acid-sensing ion channel 1 (asic1): Implication for a symmetric conformation. Cell Res., 2009, 19(8): 1035-1037. |
| Cited By in Cnki | |
| [100] | Zhang Q S, Zhou P Z, Chen Z X, et al. Dynamic pip2 interactions with voltage sensor elements contribute to kcnq2 channel gating. Proc. Natl. Acad. Sci. USA, 2013, 110(50):20093-20098. |
| Cited By in Cnki | |
| [101] | Chen L P, Zhang Q S, Qiu Y G, et al. Migration of pip2 lipids on voltage-gated potassium channel surface influences channel deactivation. Sci. Rep., 2015, 5: 15079. |
| Cited By in Cnki | |
| [102] | Cang X H, Du Y, Mao Y Y, et al. Mapping the functional binding sites of cholesterol in beta(2)-adrenergic receptor by longtime molecular dynamics simulations. J. Phys. Chem. B, 2013,117(4): 1085-1094. |
| Cited By in Cnki | |
| [103] | Cang X H, Yang L L, Yang J, et al. Cholesterol-beta(1)ar interaction versus cholesterol-beta(2)ar interaction. Proteins,2014, 82(5): 760-770. |
| Cited By in Cnki | |
| [104] | Bouyssou T, Hoenke C, Rudolf K, et al. Discovery of olodaterol, a novel inhaled beta(2)-adrenoceptor agonist with a 24 h bronchodilatory efficacy. Bioorg. Med. Chem. Lett., 2010, 20(4):1410-1414. |
| Cited By in Cnki | |
| [105] | Curreli F, Zhang H T, Zhang X H, et al. Virtual screening based identification of novel small-molecule inhibitors targeted to the hiv-1 capsid. Bioorg. Med. Chem., 2011, 19(1): 77-90. |
| Cited By in Cnki | |
| [106] | Chen H F, Chan B K, Drummond J, et al. Discovery of selective lrrk2 inhibitors guided by computational analysis and molecular modeling. J. Med. Chem., 2012, 55(11): 5536-5545. |
| Cited By in Cnki | |
| [107] | Lane J R, Chubukov P, Liu W, et al. Structure-based ligand discovery targeting orthosteric and allosteric pockets of dopamine receptors. Mol. Pharmacol., 2013, 84(6): 794-807. |
| Cited By in Cnki | |
| [108] | Lecka J, Ben-David G, Simhaev L, et al. Nonhydrolyzable atp analogues as selective inhibitors of human npp1: A combined computational/experimental study. J. Med. Chem., 2013, 56(21):8308-8320. |
| Cited By in Cnki | |
| [109] | Brown L M, Rogers K E, Aroonsakool N, et al. Allosteric inhibition of epac computational modeling and experimental validation to identify allosteric sites and inhibitors. J. Biol. Chem., 2014, 289(42): 29148-29157. |
| Cited By in Cnki | |
| [110] | Link J O, Taylor J G, Xu L H, et al. Discovery of ledipasvir (gs-5885): A potent, once-daily oral ns5a inhibitor for the treatment of hepatitis c virus infection. J. Med. Chem., 2014, 57(5): 2033-2046. |
| Cited By in Cnki | |
| [111] | Wu Y R, He C, Gao Y, et al. Dynamic modeling of human5-lipoxygenase-inhibitor interactions helps to discover novel inhibitors. J. Med. Chem., 2012, 55(6): 2597-2605. |
| Cited By in Cnki | |
| [112] | Li X, Zuo Y Y, Tang G H, et al. Discovery of a series of 2,5-diaminopyrimidine covalent irreversible inhibitors of bruton's tyrosine kinase with in vivo antitumor activity. J. Med. Chem., 2014, 57(12): 5112-5128. |
| Cited By in Cnki | |
| [113] | Meng F W, Cheng S F, Ding H, et al. Discovery and optimization of novel, selective histone methyltransferase set7 inhibitors by pharmacophore- and docking-based virtual screening. J. Med. Chem., 2015, 58(20): 8166-8181. |
| Cited By in Cnki | |
| [114] | Wang J, Luo C, Shan C L, et al. Inhibition of human copper trafficking by a small molecule significantly attenuates cancer cell proliferation. Nat. Chem., 2015, 7(12): 968-979. |
| Cited By in Cnki | |
| [115] | Li P, Chen Z X, Xu H Y, et al. The gating charge pathway of an epilepsy-associated potassium channel accommodates chemical ligands. Cell Res., 2013, 23(9): 1106-1118. |
| Cited By in Cnki | |
| [116] | Sheng C Q, Dong G Q, Miao Z Y, et al. State-of-the-art strategies for targeting protein-protein interactions by small-molecule inhibitors. Chem. Soc. Rev., 2015, 44(22): 8238-8259. |
| Cited By in Cnki | |
| [117] | Wanner J, Fry D C, Peng Z W, et al. Druggability assessment of protein-protein interfaces. Future Med. Chem., 2011, 3(16):2021-2038. |
| Cited By in Cnki | |
| [118] | Christ F, Voet A, Marchand A, et al. Rational design of smallmolecule inhibitors of the ledgf/p75-integrase interaction and hiv replication. Nat. Chem. Biol., 2010, 6(6): 442-448. |
| Cited By in Cnki | |
| [119] | Li H M, Xiao H, Lin L, et al. Drug design targeting proteinprotein interactions (ppis) using multiple ligand simultaneous docking (mlsd) and drug repositioning: Discovery of raloxifene and bazedoxifene as novel inhibitors of il-6/gp130 interface. J. Med. Chem., 2014, 57(3): 632-641. |
| Cited By in Cnki | |
| [120] | Chen Z Y, Hu Y T, Hong J, et al. Toxin acidic residue evolutionary function-guided design of de novo peptide drugs for the immunotherapeutic target, the kv1.3 channel. Sci. Rep.,2015, 5: 9881. |
| Cited By in Cnki | |
| [121] | Kong X Q, Chen L M, Jiao L Y, et al. Astemizole arrests the proliferation of cancer cells by disrupting the ezh2-eed interaction of polycomb repressive complex 2. J. Med. Chem.,2014, 57(22): 9512-9521. |
| Cited By in Cnki | |
| [122] | Pei J F, Yin N, Ma X M, et al. Systems biology brings new dimensions for structure-based drug design. J. Am. Chem. Soc., 2014, 136(33): 11556-11565. |
| Cited By in Cnki |